Computergestützte Chemie und biomolekulare Simulation
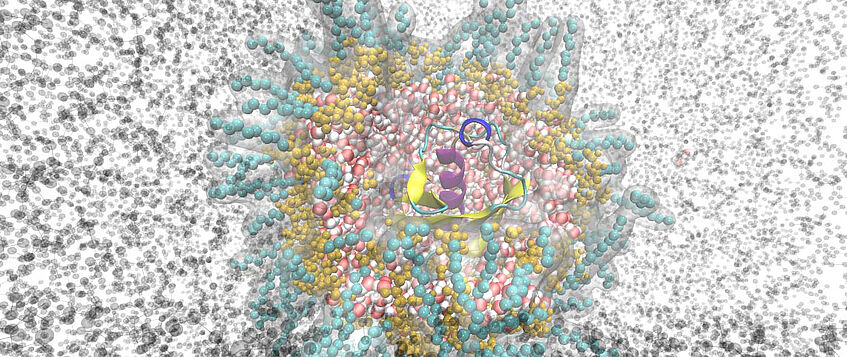
Zelluläre Nachahmung einer inversen Mizelle (Copyright: Philipp Honecker)
Die Theoretische Chemie trägt zur Charakterisierung von Eigenschaften von Materialien, sowohl im Bereich der biologischen als auch der Materialchemie, und zum Verständnis der Photochemie von organischen und anorganischen Molekülen bei. Um ein Verständnis von Strukturen, spektroskopischen Daten und Reaktivität von Molekülen zu gewinnen, werden quantenchemische Programmpakete angewandt. Die Anwendung hochgenauer Methoden zur Berechnung von Elektronenstrukturen und die Entwicklung von neuen Methoden aus dem Bereich der molekularen Reaktionsdynamik sowie die Verknüpfung dieser beiden Bereiche hat das Ziel, ein grundlegendes Verständnis von chemischen Prozessen und Struktur-Funktionszusammenhängen zu gewinnen sowie diese in Molekülen, biologischen Systemen und in Materialien quantitativ vorhersagen zu können.
Im Rahmen der Biomolekularen Simulation soll die Struktur, Dynamik und Energetik von Biopolymeren unter expliziter Behandlung der Solvatation durch konventionelle wie innovative Lösungsmittel wie zum Beispiel ionische Flüssigkeiten untersucht werden. Ziel ist hier die Analyse von Struktur und Dynamik sowohl einzelner Biomoleküle und ihrer Solvatation als auch die von Protein-Ligand- und Protein-Protein-Wechselwirkungen. Die Verbindung zu experimentellen Methoden ergibt sich über Freie-Energie-Rechnungen und die computergestützte Spektroskopie der Kernbewegung.
Die Modellierung der Struktur von Biopolymeren und ihrer Funktion in zellulären Netzwerken bildet einen weiteren Fokus. Insbesondere werden Sekundär- und Tertiärstrukturen von RNA-Molekülen, unter Einbeziehung moderner Hochdurchsatzdaten vorhergesagt. Methoden zum Design funktioneller RNA-Moleküle werden entwickelt und genutzt, um (bio)chemische Reaktionsnetzwerke zu analysieren und zu manipulieren. Die Entwicklung neuer Algorithmen profitiert von der engen Kooperation mit der Fakultät für Informatik.